
Диссертация фиброз печени у крыс
Процессы восстановления в печени у млекопитающих часто протекают по механизму заместительной или атипичной регенерации, которая также именуется фиброзом. Она сопровождается замещением функциональной ткани соединительнотканными волокнами. Фиброз возникает в ответ на повреждение печени и обратим лишь на начальных этапах. На заключительных этапах фиброз является причиной 90 % летальных исходов при хронических заболеваниях печени. В связи с этим поиск путей терапии фиброза печени является актуальной медицинской и социальной проблемой [8].
Структурные изменения в печени начинаются с изменения состава и чрезмерного накопления белков внеклеточного матрикса (ВКМ). Например, формирование фиброзных септ обусловлено увеличением синтеза коллагена, в основном клетками Ито, и связано с активацией клеток Купфера, которые усиливают экспрессию фибриногенных цитокинов и факторов роста. Активация звездчатых клеток приводит к накоплению соединительнотканного матрикса, что влечет за собой потерю микроворсинок гепатоцитами и нарушение их функции. Фиброгенез характеризуется угнетением ВКМ-деградирующих ферментов – матриксных металлопротеиназ. Таким образом, при фиброзе происходит функциональное изменение практически всех клеточных популяций печени [10, 12].
Для моделирования фиброза печени в эксперименте применяют четыреххлористый углерод (CCl4), который, действуя специфически на гепатоциты, вызывает их некроз и жировую дистрофию [13, 15].
Существует несколько подходов к терапии фиброза печени: лечение основного заболевания, ослабление воспалительного и иммунного ответа, торможение активации клеток Купфера, нейтрализация ответа звездчатых клеток, стимуляция апоптоза звездчатых клеток, усиление деградации ВКМ [8].
Ранее была изучена группа мембранотропных гомеостатических тканеспецифических биорегуляторов (МГТБ), которые в сверхмалых дозах оказывали влияние на восстановительные процессы в различных тканях [5]. Эти биологически активные вещества были выделены в отдельную группу на основании общности физико-химических свойств и характера биологического действия. Они характеризуется наличием тканевой, но не видовой специфичности и представляют собой пептидно-белковые комплексы, локализованные внеклеточно. Было показано, что МГТБ могут влиять практически на все биологические процессы: адгезия, миграция, направленность и темп дифференцировки, апоптоз клеток, а также они способны стимулировать клеточные источники регенерации, таким образом восстанавливая нормальные функцию и строение поврежденной ткани [5].
Целью данной работы явилось изучение влияния МГТБ, выделенных из печени и сыворотки крови млекопитающих на регенерацию печени мыши на фоне ее токсического повреждения CCl4. В работе описана характеристика двух пептидно-белковых комплексов – МГТБ, выделенных из сыворотки крови и печени млекопитающих, а также было изучено их влияние на развитие индуцированного фиброза печени у мышей.
Материалы и методы исследования
Для выделения биорегулятора из печени использовали крыс Wistar обоего пола, весом 220–260 г. Содержание и манипуляции с животными проводили в соответствии с директивой 86/609/EEC по обращению человека с лабораторными животными. После декапитации животных под эфирным наркозом печень перфузировали через портальную вену 10 мл раствора Рингера (0.15 М NaCl; 1 мM CaCl2; 5 мM KCl; 1 мM HEPES), удаляя основную массу крови. Печень извлекали, разрезали на фрагменты размером приблизительно 1,5–2,0 см 3 и помещали на 2–2,5 ч в р-р Рингера при 4–8 °С. Тканевой экстракт центрифугировали и далее осаждали белки сульфатом аммония (Реахим, Россия). Биорегулятор сыворотки крови получали из сыворотки крови крупного рогатого скота (БиолоТ, Россия).
К сыворотке крови и экстракту печени крыс добавляли при перемешивании сульфат аммония (780 г/л), образовавшийся раствор оставляли на 5 суток при 4 °С, центрифугировали (12000 g; 30 мин), супернатанты и осадки собирали отдельно и диализовали против воды до полного удаления следов соли. Фракции супернатантов концентрировали в вакуумном роторном испарителе при 40 °С.
Электрофорез в 12,5 %-м полиакриламидном геле проводили в денатурирующих условиях по методу Лэмли (толщина геля – 0,75 мм, размер 8х10 см). Окрашивание гелей проводили с помощью красителя Кумасси G-250 (Sigma, Германия) [11].
Исследование пептидно-белковых комплексов осуществляли на времяпролетном MALDI-TOF масс-спектрометре UltraFlex 2 (Bruker, Германия), оснащенном азотным лазером 337 нм с частотой импульса до 20 Гц. Все измерения проводили в линейном и рефлекторном режиме, определяя положительные ионы. Для накопления масс-спектров мощность лазерного излучения устанавливали на уровне минимального порогового значения, достаточного для десорбции-ионизации образца. Внешнюю калибровку проводили с использованием точных значений молекулярных масс известных белков. Образец наносили на три ячейки планшета, для каждой из которых записывали спектр, полученный в результате суммирования 10 серий спектров по 50 импульсов лазера для каждой. Для записи, обработки и анализа масс-спектров использовали программное обеспечение (Bruker, Германия): flexControl 2.4 (Build 38) и flexAnalysis 2.4 (Build 11). Точность измерения масс составляла ± 2 Да. В качестве матрицы использовали насыщенный раствор α-циано-4-гидроксикоричной кислоты (Sigma, Германия) в смеси 50 %-го ацетонитрила и 2,5 %-й трифторуксусной кислоты. Все использованные реактивы, включая воду, были аналитической или специальной для масс-спектрометрии чистоты.
Подготовка образцов, содержащих белки-модуляторы, для масс-спектрометрии проводилась следующим образом: на мишени смешивали по 0,5 мкл раствора образца и раствора 2,5-дигидроксибензойной кислоты (Sigma, Германия) 20 мг/мл в 20 %-м ацетонитриле в воде с 0,5 % ТФУ, полученную смесь высушивали на воздухе.
Масс-спектры были получены на MALDI-времяпролетном масс-спектрометре Ultraflex II (Bruker, Германия), оснащенном УФ-лазером (Nd) в режиме положительных ионов с использованием рефлектрона и в тандемном режиме (MS/MS); точность измеренных масс после докалибровки по пикам автолиза трипсина составляла 0,005 % (50 ррм), точность измеренных моноизотопных масс фрагментов была в пределах 2 Да.
Триптический гидролиз белка в полиакриламидном геле проводили следующим образом: кусочек геля размером около 2×2 мм дважды промывали для удаления красителя в 100 мкл 40 %-го раствора ацетонитрила в 0,1 М NH4HCO3 в течение 20 мин при комнатной температуре. После удаления раствора для дегидратации геля добавляли 100 мкл ацетонитрила. Удалив ацетонитрил и высушив кусочек геля, прибавляли к нему 4 мкл раствора модифицированного трипсина (Promega, США) в 0,05 М NH4HCO3 с концентрацией 15 мкг/мл. Гидролиз проводили в течение 18 ч при 37 °С, затем к раствору добавляли 7 мкл 0,5 % ТФУ в 10 %-м растворе ацетонитрила в воде и тщательно перемешивали. Раствор, образующийся над гелем, использовали для получения MALDI-масс-спектров.
Идентификацию белков осуществляли при помощи программы Mascot (www.matrixscience.com). Поиск проводился в базе данных NCBI среди белков всех организмов с указанной точностью с учетом возможного окисления метионинов кислородом воздуха и возможной модификации цистеинов акриламидом. Кандидатные белки, имеющие параметр достоверности score > 83 в базе данных NCBI считали определенными надежно (p -12 мг белка/мл (группа № 2); мыши, получавшие внутрибрюшинные инъекции CCl4 и питьевой раствор биорегулятора, выделенного из сыворотки крови крупного рогатого скота, в концентрации, соответствующей 10 -12 мг белка/мл (группа № 3); мыши, получавшие внутрибрюшинные инъекции CCl4 и Эссенциале Форте (Sanofi-Aventis, Франция) в виде питьевого раствора в концентрации 1,5 мг/мл, согласно рекомендации производителя (группа № 4); интактный контроль (группа № 5); мыши, получавшие внутрибрюшинные инъекции масла (группа № 6). Общее количество мышей в каждой экспериментальной группе было 10 голов.
Растворы биорегуляторов приготавливали путем последовательного 10-кратного разбавления исходного комплекса с концентрацией 0,1 мг/мл. Концентрацию белка в исходном комплексе определяли спектрофотометрически [1].
Для проведения гистологического исследования печени мыши проводили забор биологического материала до начала эксперимента (контроль), на 30-е и 60-е сутки эксперимента. Животных выводили из эксперимента следующим образом: после предварительной наркотизации проводили дислокацию шейных позвонков. Забор гистологического материала проводили от всех долей печени, фиксировали в ФСУ (формалин (Serva, Германия): спирт (Serva, Германия): уксусная кислота (Serva, Германия), в соотношении 9:6:1). Дальнейшую обработку материала проводили по стандартной методике. Окраску полученных срезов проводили по сложному многокрасочному методу Маллори (Serva, Германия), позволяющему специфично окрасить коллаген. Полученные гистологические препараты анализировали с использованием световой микроскопии на микроскопе Leica DM RXA2 (Leica, Германия).
Для проведения качественной оценки степени развития фиброза использовали шкалу METAVIR. Оценка проводится по развитию септ в порто-портальном и порто-центральном направлениях (F-fibrosis; F0-F4): F0 – фиброз отсутствует; F1 – звездчатое расширение портальных трактов без образования септ; F2 – расширение портальных трактов с единичными порто-портальными септами; F3 – многочисленные порто-портальные септы без цирроза; F4 – цирроз (формирование ложных долек) [2].
Результаты исследования и их обсуждение
В настоящей работе были изучены 2 представителя группы МГТБ, представляющие собой пептидно-белковые комплексы фракций супернатантов, полученные из сыворотки крови и экстракта печени. Согласно разработанному подходу [5] исследуемые комплексы были охарактеризованы с помощью ряда физико-химических и биологических методов. Было показано, что в данных комплексах присутствуют как биологически активные пептиды, так и белки, модулирующие их активность – белки-модуляторы.
Методом электрофореза в полиакриламидном геле было показано, что в обоих комплексах присутствуют белки, с мол. массами приблизительно 66000 Да, которые соответствуют сывороточному альбумину (рис. 1).
Ранее было показано, что белком-модулятором для биорегулятора, выделенного из сыворотки крови, является изоформа сывороточного альбумина, молекулярная масса которого составила 67855 Да [3]. Молекулярная масса зрелого сывороточного альбумина быка составляет 66397 Да. Использование алгоритма PROPSEARCH (база данных SwissProt) показало, что белок-модулятор сыворотки крови относится к группе преальбуминов сыворотки крови млекопитающих. Физиологическая функция преальбуминов до сих пор остается недостаточно ясной [6]. При «созревании» молекулы бычьего сывороточного альбумина происходит протеолитическое отщепление фрагмента из 23 аминокислотных остатков с N-концевой части молекулы преальбумина. Молекула преальбумина обладает сформировавшейся вторичной структурой, которая характеризуется высоким содержанием α-спиралей (около 80 %). Высокое содержание α-спиралей (79 %) было выявлено и в структуре молекулы модулятора сывороточного и печеночного биорегулятора методом кругового дихроизма: на это указывало наличие характерных минимумов при длинах волн 208 и 222 нм, соответственно [9]. Также вторичная структура белка-модулятора представлена β – складчатой структурой (2 %) и статистическим клубком (19 %).
Ранние данные по результатам секвенирования показали, что N-концевая аминокислотная последовательность молекулы сывороточного модулятора содержит 10 аминокислотных остатков и отличается от N-концевого мотива молекулы бычьего сывороточного альбумина: Ала-Про-Лиз-Лиз-Сер-Глу-Иле-Ала-Лей-Фен [3].
Для более точного определения молекулярных масс изоформ сывороточного альбумина в комплексе супернатанта печени был применен метод масс-спектрометрии после предварительного проведения триптического гидролиза. Расчетная молекулярная масса, согласно пептидам триптического гидролизата для изоформы сывороточного альбумина, выделенного из печени составила 68674 Да. В базе данных uniprot название белка gi|55628 соответствует белку семейства сывороточных альбуминов, находится в плазме крови rattus norvegicus. Соответственно, исходя из этого, белки-модуляторы, полученные из сыворотки крови и печени представляют собой малоизученные изоформы сывороточного альбумина.
Исследование комплексов фракций супернатантов печени и сыворотки крови методом MALDI-TOF масс-спектрометрии показало, что в них присутствуют небольшие пептиды (таблица).
Исследование комплексов фракций супернатантов печени и сыворотки крови методом кругового дихроизма показало преобладание во вторичной структуре изучаемых комплексов β-структур и статистического клубка (таблица).
Исходя из данных метода кругового дихроизма, наличие незначительного количества спиралей указывает на присутствие в комплексах небольшого количества изоформ альбумина. Методом лазерного динамического светорассеяния было показано наличие крупных наноразмерных частиц в водных растворах МГТБ печени (100 ± 4,6 нм) и сыворотки крови (62,05 ± 3,0 нм) соответственно.
Далее оба МГТБ исследовали на модели индуцированного фиброза у млекопитающих. Исследование показало, что МГТБ, выделенные из печени и сыворотки крови, по-разному влияют на его развитие.
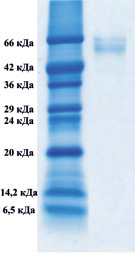
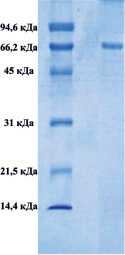
Рис. 1. Электрофорез в 12,5-м % полиакриламидном геле комплекса супернатанта тканевого экстракта печени крысы (А), комплекса супернатанта сыворотки крови (Б)
Физико-химические свойства исследуемых комплексов
Источник
Фиброз печени как показание для терапии у больных хроническими гепатитами
- КЛЮЧЕВЫЕ СЛОВА: фиброз печени, хронический гепатит, глицирризиновая кислота, Фосфоглив
Актуальность проблемы фиброза печени
Целесообразность оценки фиброза печени (ФП) у больных хроническими гепатитами по-прежнему вызывает вопросы у практикующих врачей. Это обусловлено тем, что данная патология не имеет специфических клинических проявлений и не доставляет пациентам особого беспокойства. Тем не менее наличие ФП позволяет установить прежде всего стадию хронического гепатита. Повторное определение стадии ФП при хроническом гепатите актуально для прогнозирования, уточнения темпов прогрессирования заболевания и скорости развития цирроза печени (ЦП).
Последние десятилетия проблеме ФП было посвящено множество клинических и экспериментальных исследований, предприняты попытки стандартизировать правила ведения пациентов с продвинутым фиброзом и ЦП, определена тактика применения ряда патогенетических препаратов. Между тем в практике терапевта ФП не рассматривается как отдельное показание для лечения, что существенно отличается от опыта лечения хронических заболеваний печени инфекционистами, у которых это показание считается стандартным.
Актуальность обозначенной проблемы обусловлена также высокой смертностью от терминальной стадии ФП – цирроза, который занимает 9-е место среди всех причин смерти в мире и 6-е место – среди лиц наиболее трудоспособного возраста – от 14 до 30 случаев на 100 000 населения. В нашей стране этот показатель значительно выше – до 60 случаев на 100 000 населения [1].
Прогностически неблагоприятным у больных с выраженными стадиями ФП является наличие высокого риска развития гепатоцеллюлярной карциномы. Этот риск более чем в 30 раз превышает таковой у пациентов с начальным ФП.
Таким образом, совершенствование ранней диагностики ФП, оценка темпов его прогрессирования и разработка методов коррекции процессов фиброгенеза в печени – чрезвычайно важные задачи не только для гепатологии, но и для внутренних болезней в целом.
Механизмы формирования ФП
Формирование ФП – один из универсальных вариантов морфологической реакции печени на повреждение. Вообще на повреждение печень реагирует такими формами внутренних трансформаций, как стеатоз, пигментные отложения, тромбоз, апоптоз, некроз, адаптация, пролиферация гепатоцитов и собственно фиброзные изменения. Важно также и то, что независимо от этиологического фактора и типа морфологического реагирования только выраженность фиброгенеза позволяет определить стадию заболевания. Именно стадия ФП отражает темпы прогрессирования хронических гепатитов.
Ранее считалось, что ФП – это процесс, направленный исключительно в сторону ЦП. Сегодня такая точка зрения пересмотрена. Изучается возможность регрессии ЦП, что предполагает терапевтические перспективы воздействия на подобный вариант реагирования гепатоцитов [2–4].
Исходя из современных представлений, эволюция фиброза в ЦП – это не просто чередование процессов повреждения и рубцевания в печени. Интересно, что терапевтические подходы к лечению ФП на данном этапе связывают исключительно с этиотропным воздействием. Это один из самых важных временных промежутков в течении хронического заболевания печени, при котором в целом понятны процессы воспаления (некроза) печени, замещения поврежденных гепатоцитов соединительной тканью, собственно фиброгенез и реакция сосудистого русла печени, то есть формирование портальной гипертензии (ПГ) и ЦП. Однако ЦП в данной модели представляется как простое следствие выраженного фиброгенеза и накопление большого количества соединительной ткани. Что касается определения понятия ЦП, его рассматривают как хронический процесс, приводящий к развитию ложных долек, то есть к изменению цитоархитектоники органа с возникновением нового сосудистого русла [5]. Следовательно, ведущим фактором формирования ЦП является не столько рост соединительной ткани, сколько ангиогенез. В экспериментальных исследованиях показано, что распространенность и выраженность ФП коррелируют с сосудистым показателем портальной гипертензии HVPG (hepatic venous pressure gradient – градиент венозного давления в печени) [6, 7].
Значит, можно говорить о коррекции известной патогенетической цепочки событий в прогрессировании хронического гепатита, включающей пять этапов.
I. Повреждение/воспаление гепатоцитов.
II. Изменение взаимодействия клеток синусоидального компартмента, стимуляция синусоидального фиброгенеза и формирование синусоидальной (функциональной) ПГ.
III. Стимуляция печеночного неоангиогенеза, развитие системной сосудистой реакции с разобщением печеночного и системного кровотока.
IV. Формирование ложных долек и компенсированного ЦП.
V. Формирование органической ПГ и декомпенсация ЦП.
Иными словами, выраженность фиброза и размеры узлов-регенератов в печени отражают конечную стадию процесса и служат предикторами клинически значимой ПГ (давление в системе портальных вен выше 10 мм рт. ст.), с которой по сути и связан окончательный прогноз заболевания.
Механизмы повреждающего воздействия различных этиологических факторов, а также заключительный этап развития выраженной ПГ и декомпенсации ЦП подробно изложены в работах отечественных и зарубежных авторов [5, 7, 8]. В данной статье рассматриваются промежуточные этапы (II и III) прогрессирования хронических заболеваний печени как наиболее перспективные для терапевтического воздействия на процессы фиброгенеза.
Изменение взаимодействия клеток синусоидального компартмента, стимуляция синусоидального фиброгенеза и формирование синусоидальной (функциональной) ПГ
Взаимодействие в синусоидальном компартменте включает закономерные изменения интимных межклеточных механизмов между гепатоцитами, клетками Ито, макрофагами Купфера и эндотелиоцитами (см. рисунок). В результате воздействия этиологических факторов и повреждения гепатоцитов выделяется большое количество цитокинов. Наиболее важный из них фактор некроза опухоли (ФНО) альфа [9–11].
В свою очередь макрофаги под воздействием этиологических факторов осуществляют фагоцитоз бактерий и вирусов, связывание и удаление иммуноглобулина (Ig) G и липосахаридов, выделение вазоактивных субстанций, влияющих на функциональную активность клеток Ито и эндотелиоцитов. Из последних особый интерес представляет фактор агрегации тромбоцитов (transforming growth factor beta 1 – TGF-бета-1) [12].
Эндотелиальные клетки печени играют роль мини-сфинктеров, не имеющих базальной мембраны. Их структура представлена касающимися друг друга боковыми контактами клетками, образующими своеобразную сеть или сито, через которые проходят вещества определенной молекулярной массы. Они также осуществляют захват гиалуроновой кислоты, связывание IgG, продукцию цитокинов, клеточный эндоцитоз и пиноцитоз.
Существенную роль в фиброгенезе играют клетки Ито, или полипотентные звездчатые клетки (ПЗК), открытые в 1970-х гг. В норме они осуществляют накопительную функцию, депонируя липопротеины высокой плотности и витамин А. Однако, подвергаясь воздействию таких вазоактивных веществ, как ФНО-альфа, TGF-бета-1, цитокины, хемокины, интегрины, ПЗК способны трансформироваться в переходные клетки и далее в миофибробласты, отростки которых распространяются за пределы одного синусоида. Эти клетки регулируют синусоидальный кровоток как напрямую – путем изменения собственной сократительной способности, так и опосредованно – через изменение синтеза металлопротеиназ и их ингибиторов, задействованных в образовании проколлагена [13]. То есть специфические цитокины, вырабатываемые клетками синусоидального компартмента и регулирующие воспалительный ответ на повреждение гепатоцитов, управляют фиброгенезом, не только способствуя трансформации ПЗК в миофибробластоподобные клетки, но и стимулируя синтез ими экстрацеллюлярного коллагенового матрикса (ЭКМ) и ингибируя его
деградацию.
Способность миофибробластов сокращаться и изменять просвет синусоида обусловливает функциональный (преходящий) компонент ПГ. Последующее угнетение активности металлопротеиназ с преобладанием их ингибиторов приводит к накоплению коллагена в пространстве Диссе, что отражает механизмы синусоидального фиброгенеза.
Стимуляция печеночного неоангиогенеза, развитие системной сосудистой реакции с разобщением печеночного и системного кровотока
Прогрессирование синусоидального фиброгенеза приводит к формированию базальной мембраны у эндотелиоцитов и нарушению тока крови к синусоидальному полюсу гепатоцита. Это отражает переход физиологической реакции организма, направленной на уменьшение антигенного притока крови к печеночным клеткам, в патофизиологическую, то есть полное прекращение доступа крови с последующим ростом синусоидального давления и прогрессированием ПГ. Именно такая совокупность последовательных изменений способствует развитию системной реакции организма в виде выработки стимуляторов неоангиогенеза и разобщения печеночного и системного кровотока. При этом в печеночной крови начинают преобладать такие вазоконстрикторы, как эндотелины 1 и 2, адреналин, ангиотензин II, вазопрессин, а в системном кровотоке – вазодилататоры (оксид азота, оксид углерода, простациклин, глюкагон, простагландин Е2, предсердный натрийуретический гормон).
Вазодилатирующие субстанции, такие как оксид и нитрит азота, релаксин, обладают еще и антифибротическими свойствами. В то же время вазоконстриктору ангиотензину II, по данным ряда авторов, принадлежит основная роль в стимуляции фиброгенеза печени. Мощный вазоконстриктор эндотелин 1 также стимулирует темпы фиброзных изменений в печени, воздействуя на рецепторы типа А [5, 11].
Разобщение печеночного и системного кровотока сопровождается параллельной стимуляцией симпатоадреналовой системы (САС) и изменением соотношения гуморально-метаболических факторов с активацией ренин-ангиотензин-альдостероновой системы (РААС).
Компенсаторная реакция сердечно-сосудистой системы включает формирование гиперкинетического типа центральной гемодинамики с ростом минутного объема кровотока, что в свою очередь также может приводить к повышению давления в воротной вене за счет усиления портального кровотока [5, 14].
Дальнейшая стимуляция неоангиогенеза вследствие нарушения равновесия между тонкими механизмами, влияющими на фиброгенез, и связанное с ним состояние кровотока в воротной вене способствуют формированию нового сосудистого русла печени, органической ПГ и соответственно компенсированного ЦП.
Ряд других печеночных клеток, таких как миофибробласты из мелких портальных сосудов, также могут иметь фиброз-продуцирующую способность, которая активируется вокруг портальных сосудов и билиарного тракта печени при нарушении желчеоттока. Как следствие – дополнительное накопление коллагена. Наличие синдрома холестаза является независимым фактором стимуляции фиброгенеза, что принципиально ускоряет прогрессирование заболевания печени.
Таким образом, в настоящее время патогенез ФП рассматривается как эволюционный процесс, включающий не только внутрипеченочные, но и системные механизмы.
На начальном, внутрипеченочном этапе происходит изменение взаимодействия клеток синусоидального компартмента, инициация синусоидального фиброгенеза и формирование синусоидальной (функциональной) ПГ.
Последующий дисбаланс вазоактивных субстанций со стимуляцией печеночного неоангиогенеза приводит к развитию системной сосудистой реакции с разобщением печеночного и системного кровотока, а также вовлечением в процесс САС и РААС. Реактивные механизмы, протекающие на уровне целостного организма, в сочетании с внутрипеченочными патогенетическими изменениями усугубляют нарушение кровоснабжения и повреждение печени и способствуют дальнейшему прогрессированию процессов фиброгенеза.
В этой связи важна оценка предикторов активного фиброгенеза, к которым относятся не только повреждающие агенты и факторы, стимулирующие рост соединительной ткани, но и причины, усугубляющие указанные выше этапы патогенетического процесса [15]. Так, к предикторам активно прогрессирующего ФП могут быть отнесены:
- исходное и приобретенное наличие нарушений желчеоттока (холестаз);
- возраст старше 45 лет, поскольку к этому моменту высок естественный уровень накопления соединительной ткани в печени;
- мужской пол как фактор более высокого уровня алкоголизации и риска ПГ;
- высокая вирусная нагрузка при НВV-инфекции (хронический гепатит B);
- синдромы перегрузки железом и медью;
- злоупотребление алкоголем;
- инсулинорезистентность, абдоминальное ожирение, жировой гепатоз;
- сопутствующие заболевания сердечно-сосудистой системы, в том числе сердечная недостаточность.
Методы оценки ФП
Методы оценки фиброза подразделяются на лабораторные и инструментальные.
Лабораторные методы оценки ФП
Лабораторные методы включают оценку прямых и непрямых маркеров фиброза. К прямым относятся методики количественного измерения показателей ЭКМ или маркеров его метаболизма: гиалуроновой кислоты, ламинина, проколлагеновых пептидов, типов коллагенов, матриксных металлопротеиназ и их ингибиторов.
Непрямыми маркерами фиброза считаются традиционные сывороточные показатели воспалительного процесса в печени, а именно уровни аминотрансфераз, коэффициент Де Ритиса, уровни гамма-глутамилтранспептидазы, аполипопротеина А1, альфа-2-макроглобулина, гаптоглобина, количество тромбоцитов.
В настоящее время наиболее актуальны непрямые маркеры ФП, объединенные в комплексные тесты методом дискриминантного анализа (ФиброАктиТест, ФиброМаксТест, СтеатоСкрин и др.) [16].
Инструментальные методы оценки ФП
Визуализирующие методы обследования. Широко распространенные и доступные методы визуального неинвазивного обследования (ультразвуковая диагностика и компьютерная томография) могут применяться для установления диагноза ФП, признаков ПГ, ряда сосудистых нарушений (допплерография сосудов портальной системы). Однако их диагностическая информативность на ранних стадиях фиброза низкая.
Ультразвуковая эластография (УЗИ-ЭГ) – новейший метод обследования внутренних органов, позволяющий получить информацию о жесткости тканей печени. Сначала УЗИ показывает общее состояние органа, выявляет подозрительные участки, осуществляет так называемый пристрел. Далее на этих участках применяется УЗИ-ЭГ. С ее помощью врач получает подробную информацию о локальных свойствах ткани. Метод заключается в том, что к ультразвуку добавляют давление. В сжатом состоянии ткани различной плотности по-разному проводят ультразвук. Аппарат с возможностью эластографии фиксирует эту разницу, на основании чего определяет плотность тканей. Преимуществами метода являются высокая скорость получения результата и отсутствие противопоказаний, в том числе возможность проведения исследования при беременности. УЗИ-ЭГ печени не только характеризует общее состояние органа, но и выявляет наличие и стадию фиброзных изменений.
Фиброэластография печени аппаратом «ФиброСкан» (ФЭГ). Это неинвазивная технология визуализации неоднородностей мягких тканей. В процессе данного исследования на изучаемую область оказывают дополнительное давление. Из-за неодинаковой эластичности неоднородные элементы ткани сокращаются по-разному. Скорость распространения упругих волн определяется эластичностью печеночной ткани, что позволяет выявлять патологические участки в органе. Косвенная инструментальная оценка выраженности фиброза посредством измерения эластичности печени с помощью аппарата основана на генерации низкочастотных колебаний, передающихся на ткань печени. Методика основана на использовании колебаний низкой частоты для количественной оценки эластичности как показателя состояния печени и процентного содержания в ней соединительной ткани. Доказана положительная корреляционная связь между результатами, полученными при ФЭГ, и биопсией печени [17, 18].
Биопсия печени. Данная процедура позволяет диагностировать ФП с достаточно высокой степенью достоверности. В 15–35% случаев при выполнении пункционной биопсии печени получают неизмененную ткань печени, в 1,5% случаев – неинформативный материал, при том что только 5% пациентов соглашаются на проведение этого исследования [19–21].
Сравнительная оценка методов определения ФП представлена в табл. 1.
Классификация ФП, которой большинство специалистов пользуется до сих пор, была предложена на конгрессе гепатологов в 1994 г. в Лос-Анджелесе. Она рассматривает наличие фиброза и соответственно стадию заболевания:
- 0 – фиброз отсутствует;
- 1 – слабо выраженный перипортальный фиброз;
- 2 – умеренный фиброз с портопортальными септами;
- 3 – выраженный фиброз с портоцентральными септами;
- 4 – ЦП.
Последующие классификации включают изложенную выше как составную часть и имеют особенности при отдельных нозологических формах [22, 23].
Большинству врачей, использующих в повседневной клинической деятельности ФП как оценочный показатель, абсолютно понятны стадии 0 и 4. Не вызывает больших затруднений оценка 3-й стадии, которая хорошо визуализируется при УЗИ и фибросканировании. Но определение 1-й и 2-й стадий фиброза сопряжено, как правило, с отсутствием понимания значимой разницы между ними.
Выраженность фиброзных изменений в печени как критерий оценки стадии заболевания отражает дисбаланс качественных и количественных составляющих ЭКМ, включающего различные варианты соединительной ткани и коллаген 1–4-го типов. Например, не очень понятная практикующим специалистам разница между начальной (перипортальной, F1) и умеренной (портопортальной, F2) стадией фиброза подразумевает в 4–6 раз большее количество ЭКМ [10].
Принципы терапии ФП
Вопросы антифибротической терапии как самостоятельного подхода давно обсуждаются в клинической практике. Еще в 2003 г.
R. Safadi и S.L. Friedman разработали ее основные принципы, включавшие воздействие на все патогенетические звенья процесса. К основным направлениям лечения были отнесены:
- устранение этиологического фактора (табл. 2);
- устранение воспалительных изменений в печени;
- ингибирование активации ПЗК;
- подавление эффектов активированных ПЗК;
- повышение репарации тканей;
- стимуляция клеточного апоптоза.
Остальные направления в лечении ФП являются в основном перспективными и широко не применяются в клинической практике. Поэтому чрезвычайно актуальным остается поиск новых подходов к коррекции данного состояния.
Общепринятых стандартов лечения ФП не разработано, хотя существует ряд экспериментальных моделей для исследования [24, 25].
На сегодняшний день к основным подходам терапии ФП при исключении воздействия на этиологический фактор относят:
- патогенетическое лечение основного заболевания в случаях, когда на этиологический фактор воздействовать невозможно или этиология болезни не установлена;
- уменьшение активности воспалительного процесса в печени;
- торможение активации ПЗК;
- активацию механизмов фибролиза для разрушения избытка белков ЭКМ;
- воздействие на ПГ.
Лекарственные средства, используемые для терапии ФП
Все лекарственные средства для терапии ФП теоретически можно разделить на три группы:
1) пепараты, действующие на конкретные механизмы фиброгенеза;
2) лекарственные средства неспецифического действия;
3) препараты, действующие на ПГ.
К препаратам, действующим на конкретные механизмы фиброгенеза, относят:
- интерфероны и синтетические аналоги нуклеозидов при вирусных заболеваниях [26–29];
- препараты, снижающие концентрацию ФНО-альфа (пентоксифиллин, глицирризиновая кислота (ГК), бигуаниды);
- препараты, подавляющие избыточную активацию ПЗК и снижающие уровень TGF-бета-1 (урсодезоксихолевая кислота (УДХК), ГК, витамин Е, эссенциальные фосфолипиды (ЭФЛ), силимарин);
- антагонисты эндотелина (амбризентан, бозентан, ситаксентан, тезосентан);
- ингибиторы каспаз (ГК, УДХК, GS9450), ингибиторы апоптоза (ГК, УДХК, TRO19622).
Препаратами неспецифического действия считаются:
- мембраностабилизаторы и антиоксиданты (препараты янтарной кислоты, токоферол);
- флавоноиды, ЭФЛ.
Средствами, действующими на ПГ, являются бета-блокаторы, нитропрепараты, ингибиторы ангиотензинпревращающего фермента (иАПФ) и антагонисты рецепторов ангиотензина (АРА) [5, 14].
Результаты исследований последних лет в отношении ряда лекарственных средств подтверждают их антифибротическое действие, доказанное пока преимущественно на экспериментальных моделях. Одним из таких средств является ГК.
ГК как препарат, действующий на конкретные механизмы фиброгенеза, изучали еще в начале XX века японские ученые в качестве антифибротического препарата. Данные клинических исследований показали, что ГК нормализует ферментный спектр крови у больных хроническим гепатитом C [30–33].
Прямые антифибротические механизмы ГК доказаны на экспериментальной модели ФП крыс, вызванного подкожными инъекциями ССl 4 . С помощью RT-PCR-анализа (reverse transcription – polymerase chain reaction, или РТ-ПЦР-анализ) было показано увеличение экспрессии генов (smurf2, PTAFR, CYP2D6, FGG), связанных с воспалением. На основании полученных данных доказан антифибротический эффект ГК у животных [34]. Интимный механизм ингибирования процессов фиброгенеза включал торможение трансформации ПЗК в миофибробласты, восстановление активности тканевых металлопротеиназ и снижение активности их ингибиторов, регулирующих баланс между процессами синтеза и деградации ЭКМ.
Эта гипотеза была подтверждена в последующих экспериментальных и клинических исследованиях. Так, ГК подавляла активацию промотора гена коллагена COL1A2 и прогрессирование ФП, вызванного повторным введением тетрахлористого углерода. Кроме того, ГК подавляла синтез коллагена 1-го типа на уровне транскрипции гена [35].
Одним из механизмов прямого антифибротического действия ГК, зафиксированного у людей, стало подавление экспрессии РНК проколлагена типов I и III [36].
Так, у пациентов с хроническим гепатитом C под влиянием ГК отмечалось снижение активности ПЗК. Это снижение приводило к уменьшению продукции ингибиторов протеаз и металлопротеиназ с дальнейшим восстановлением активности последних и соответственно дегрануляции избыточного коллагена. При этом снижение активации ПЗК сопровождалось индукцией в них процессов апоптоза [37].
Кроме прямого антифибротического у ГК были выявлены другие дополнительные эффекты.
Как известно, благодаря своему строению ГК является синергистом глюкокортикостероидов [38]. ГК блокирует внутриклеточные сигнальные пути, реализующие воспаление, на уровне NF-kappa B [39, 40], STAT3 [41], TLR4 [40], HMGB1 [42, 43], JNK, ERK, PI3K/AKT [44], MAPKs [40]. ГК угнетает выработку ФНО-альфа, интерлейкина (ИЛ) 6 и ингибирует миелопероксидазы [39, 40]. По сути, оказывая антагонистическое влияние на воспалительный процесс, ГК опосредованно тормозит темпы фиброгенеза.
Помимо противовоспалительного действия ГК обладает антиоксидантной активностью, которую связывают с торможением перекисного окисления липидов через фосфорилирование 5-липоксигеназы. Кроме того, ГК способна связываться с прооксидантом простагландином Е2 [45].
В ряде работ продемонстрировано антиапоптотическое действие ГК, обусловленное ингибированием каспазы-3 [46] и угнетением высвобождения из митохондрий цитохрома С [39].
При изучении влияния ГК на синтез и пролиферацию ДНК в первичной культуре гепатоцитов взрослых крыс установлено, что глицирризин стимулирует фосфорилирование рецепторов эпидермального фактора роста и p42 MAP-киназы и активирует синтез ДНК гепатоцитов. Кроме того, ГК ингибирует рост опухолей у мышей за счет опосредованного противоопухолевого эффекта. Показано защитное действие ГК против индуцированного канцерогенами повреждения ДНК, а также возможность понижающей регуляции рецептора эпидермального фактора роста. Входящие в состав солодки полифенолы индуцировали апоптоз раковых клеток [36].
В ходе последующих рандомизированных исследований было показано, что в отличие от плацебо ГК приводит к значительному снижению уровня аминотрансфераз печени и улучшению гистологической картины [47]. Сведения о краткосрочной и долгосрочной эффективности данного препарата получены в ходе ретроспективного исследования, результаты которого продемонстрировали, что длительный прием ГК препятствует развитию ЦП и гепатоклеточной карциномы на фоне хронического гепатита C [48].
Фосфатидилхолин как составляющая часть ЭФЛ также обладает антифибротическим действием и способствует регрессии фиброгенеза путем подавления активности коллагеназы и трансформации ПЗК в коллагенпродуцирующие клетки, что дополняется противовоспалительным действием за счет уменьшения синтеза провоспалительных цитокинов (ФНО-альфа, ИЛ-1b). Однако доказательства антифибротического эффекта представлены только у нечеловекообразных обезьян [49]. Аналогичные исследования у людей такого эффекта не продемонстрировали [50].
ЭФЛ в комбинации с ГК улучшают ее липофильные свойства, повышая биодоступность и противовоспалительную активность [51–53]. Данный синергический эффект положен в основу разработки отечественного препарата ГК Фосфоглив, который представляет собой комбинацию высоко биодоступных ЭФЛ и ГК. На сегодняшний день Фосфоглив является одним из стандартных и универсальных средств для лечения ФП любой этиологии.
В качестве перспективных направлений антифибротической терапии в эксперименте была доказана клиническая эффективность применения факторов роста (IGF, фактор роста гепатоцитов, кардиотропин) и факторов, стимулирующих гены, ответственные за их продукцию.
Блокаторы эндотелина-1 рецепторов типа А и вазодилататоры (простагландин Е2 и донорский нитрит азота) показали хорошую антифибротическую активность у грызунов, однако у людей подобные эффекты не оценивались.
Альтернативный подход к лечению ФП заключается в применении ингибиторов коллагена и промоутеров их деградации. Так, ингибиторы пролил-4-гидроксилазы и галофуджинона предупреждают развитие экспериментального ЦП ингибированием синтеза коллагена. Такие факторы, как ММР-8 и активаторы плазминогена и урокиназы, стимулируют деградацию коллагена in vivo.
В эксперименте с инфузией мезенхимальных стволовых клеток получено уменьшение индуцированного фиброза, что потенциально может быть использовано в будущем при лечении хронических заболеваний печени [54].
В настоящее время проведено достаточное количество научных исследований, в которых подтверждена важность ингибирования РААС как фактора, влияющего на ПГ и уменьшающего темпы прогрессирования ФП, что может стать многообещающей стратегией в лечении этого состояния. Данный факт подтверждается тем, что указанная группа препаратов в качестве антифибротических агентов вошла в стандарты лечения и широко используется у пациентов с хроническими заболеваниями сердца и почек.
Согласно отчетам отделений трансплантологии, у пациентов, принимающих ингибиторы РААС и антигипертензивные препараты, после трансплантации печени прогрессия фиброза менее выражена, чем у больных, получающих другие виды лечения. Однако многоцентровые клинические исследования в настоящее время еще не завершены и результаты не обработаны.
Пентоксифиллин (ингибитор фосфодиэстеразы), амилорид (ингибитор натрий-водородной помпы), S-фарниезилтиосалициловая кислота (антагонист ренин-ангиотензиновой системы) как субстанции, ингибирующие ключевые сигнальные пути фиброгенеза печени, также имеют потенциальное значение в комплексной терапии данного состояния. Для конкретных форм поражения печени, в частности при неалкогольном стеатогепатите, воздействие на специфические лиганды, например на cнижение экспрессии гена SREPB-1c и увеличение активности PPRA-альфа (peroxisome proliferator-activated receptor), оказывает положительный эффект на регресс ФП преимущественно у пациентов с нарушениями углеводного обмена (S16).
Лечение ПГ у больных ФП следует начинать на самом раннем этапе, когда появляются первые изменения воротного кровоснабжения.
На стадии начальной ПГ гемодинамические расстройства происходят преимущественно на функциональном уровне, с развитием симпатикотонии, появлением расстройств микроциркуляции и реологических свойств крови. Для коррекции начальной ПГ рекомендуется применение бета-блокаторов как вегетокорректоров, антиагрегантов, пентоксифиллина и антиоксидантов. При умеренной и выраженной ПГ терапия дополняется антагонистами альдостерона, иАПФ, АРА [14].
По существу доступной и унифицированной схемой антифибротической терапии, особенно у пациентов с продвинутым фиброзом и ПГ, считается:
- комбинация пероральной и парентеральной форм Фосфоглива (5,0 внутривенно, 3–5 раз в неделю), курс до 12 месяцев и более;
- УДХК в дозе 15–20 мг/кг/сут, постоянный прием;
- пропранолола 40–80 мг/сут. Показателем эффективности и достаточности дозы препарата является уменьшение частоты сердечных сокращений на 25% по сравнению с исходной, постоянный прием;
- верошпирона в дозе нейрогормонального модулятора 12,5–25 мг/сут, продолжительность аналогична таковой пропранолола;
- иАПФ или АРА (показания и дозы не отработаны).
В качестве дополнительных агентов, усиливающих схему, используются:
- токоферол (100–150 мг/сут), курс до 4–8 недель;
- пентоксифиллин (2%-ный, 5 мл внутривенно капельно), лечение следует проводить курсами до 5–10 процедур.
Наличие ФП у больных хроническими гепатитами служит самостоятельным показанием для терапии, целью которой является не только снижение темпов прогрессирования процессов фиброгенеза, но и предотвращение развития ПГ, ЦП и его фатальных осложнений.
Следует учитывать необходимость комплексного подхода при составлении программы лечения ФП, включающей воздействие на этиологические факторы и патогенетические механизмы, такие как холестаз и ПГ, наличие которых ускоряет прогрессирование фиброгенеза.
Терапия должна начинаться в максимально ранние сроки. Чем меньше исходная стадия фиброза и выраженность ПГ, тем более полно возможно его обратное развитие. Несмотря на развитие ЦП, антифибротическая терапия должна быть продолжена. Пока сохраняется даже небольшая часть печеночной ткани, процесс фиброза в печени прогрессирует.
Существующий вариант универсальной и доступной схемы антифибротической терапии включает: препараты ГК, УДХК, бета-блокаторы, антагонисты альдостерона, иАПФ или АРА, микроциркулянты и антиоксиданты.
Источник